El estudio genético que te ayudará a planificar la llegada de un hijo sano.

GendaFamily
Conocé las enfermedades hereditarias que se podrían estar transmitiendo silenciosamente en tu familia.
Te ayudamos a planificar la llegada de un hijo sano.
Te ayudamos a tomar decisiones inteligentes sobre tu salud, tu familia y tu futuro.
Si estás pensando en tener hijos, conocé si vos o tu pareja son portadores de mutaciones para enfermedades hereditarias que podrían afectar a tu familia.
GendaFamily es un estudio genético que te permite determinar si sos portador asintomático de alguna enfermedad hereditaria recesiva que podrían desarrollar tus hijos.
El estudio analiza 569 genes.
| Abetalipoproteinemia | MTTP |
| Acidemia glutárica tipo I | GCDH |
| Acidemia glutárica tipo II (relacionada con ETFA) | ETFA |
| Acidemia glutárica tipo II (relacionada con ETFDH) | ETFDH |
| Acidemia isovalérica | IVD |
| Acidemia malónica y metilmalónica combinada (relacionada con ACSF3) | ACSF3 |
| Acidemia metilmalónica (relacionada con MMAA) | MMAA |
| Acidemia metilmalónica (relacionada con MMAB) | MMAB |
| Acidemia metilmalónica (relacionada con MUT) | MUT |
| Acidemia metilmalónica con homocistinuria, tipo cobalabina D | MMADHC |
| Acidemia metilmalónica con homocistinuria, tipo cobalamina C | MMACHC |
| Acidemia propiónica (relacionada con PCCA) | PCCA |
| Acidemia propiónica (relacionada con PCCB) | PCCB |
| Acidosis renal tubular con sordera (relacionada con ATP6V1B1) | ATP6V1B1 |
| Aciduria 3-metilglutacónica tipo III (atrofia óptica de Costeff) | OPA3 |
| Acrodermatitis enteropática | SLC39A4 |
| Acromatopsia (relacionada con CNGB3) | CNGB3 |
| Adrenoleucodistrofia ligada al X | ABCD1 |
| Alcaptonuria | HGD |
| Alfa talasemia | HBA1/HBA2 |
| Alfa-manosidosis | MAN2B1 |
| Amaurosis congénita de Leber 10 / Desórdenes relacionados con CEP290 | CEP290 |
| Amaurosis congénita de Leber 13 | RDH12 |
| Amaurosis congénita de Leber 2 | RPE65 |
| Amaurosis congénita de Leber 5 | LCA5 |
| Amaurosis congénita de Leber 8 / Desórdenes relacionados con CRB1 | CRB1 |
| Anemia de Fanconi tipo A | FANCA |
| Anemia de Fanconi tipo C | FANCC |
| Anemia de Fanconi tipo G | FANCG |
| Argininosuccinil aciduria | ASL |
| Aspartilglucosaminuria | AGA |
| Ataxia con déficit de vitamina E | TTPA |
| Ataxia espástica autosómica recesiva de Charlevoix-Saguenay | SACS |
| Ataxia-telangiectasia | ATM |
| Atrofia cerebral y microcefalia progresiva postnatal / Atrofia cerebral y cerebelosa infantil | MED17 |
| Atrofia muscular espinal | SMN1 |
| Cistinosis | CTNS |
| Citrulinema tipo 1 | ASS1 |
| Colestasis intrahepática progresiva congénita | ABCB11 |
| Condrodisplasia punctata rizomélica tipo 1 / enfermedad de Refsum (relacionada con PEX7) | PEX7 |
| Condrodisplasia punctata rizomélica tipo 3 | AGPS |
| Corea acantocitosis | VPS13A |
| Coroideremia | CHM |
| Deficiencia combinada de hormonas hipofisiarias (relacionada con LHX3) | LHX3 |
| Deficiencia combinada de hormonas hipofisiarias (relacionada con PROP1) | PROP1 |
| Deficiencia combinada de la fosforilación oxidativa (relacionada con GFM1) | GFM1 |
| Deficiencia combinada de la fosforilación oxidativa (relacionada con TSFM) | TSFM |
| Deficiencia de 3-hidroxi-3-metilglutaril-CoA (HMG-CoA) liasa | HMGCL |
| Deficiencia de 3-hidroxiacil-CoA deshidrogenasa de cadena larga | HADHA |
| Deficiencia de 3-metilcrotonil-CoA carboxilasa relacionada con MCC1 | MCCC1 |
| Deficiencia de 3-metilcrotonil-CoA carboxilasa relacionada con MCC2 | MCCC2 |
| Deficiencia de ACAD9 | ACAD9 |
| Deficiencia de acil-CoA deshidrogenasa (MCAD) de cadena media | ACADM |
| Deficiencia de acil-CoA deshidrogenasa de cadena muy larga (VLCAD) | ACADVL |
| Deficiencia de acil-CoA oxidasa peroxisomal | ACOX1 |
| Deficiencia de adenosina desaminasa | ADA |
| Deficiencia de alfa-1-antitripsina | SERPINA 1 |
| Deficiencia de arginasa | ARG1 |
| Deficiencia de aromatasa | CYP19A1 |
| Deficiencia de asparagina sintetasa | ASNS |
| Deficiencia de Beta-cetotiolasa | ACAT1 |
| Deficiencia de biotinidasa | BTD |
| Deficiencia de carbamilfosfato sintetasa tipo I | CPS1 |
| Deficiencia de carnitina palmitoil-transferasa I | CPT1A |
| Deficiencia de carnitina palmitoil-transferasa II | CPT2 |
| Deficiencia de carnitina primaria | SLC22A5 |
| Deficiencia de citrina | SLC25A13 |
| Deficiencia de corticosterona metiloxidasa | CYP11B2 |
| Deficiencia de factor IX (Hemofilia B) | F9 |
| Deficiencia de fenilalanina hidroxilasa (incluida fenilcetonuria (PKU)) | PAH |
| Deficiencia de fosfoglicerato deshidrogenasa / Síndrome de Neu-Laxova | PHGDH |
| Deficiencia de fumarato hidratasa | FH |
| Deficiencia de guanidinoacetato metiltransferasa | GAMT |
| Deficiencia de holocarboxilasa sintetasa | HLCS |
| Deficiencia de la dihidrolipoamida deshidrogenasa | DLD |
| Deficiencia de la glucosa-6-fosfato deshidrogenasa | G6PD |
| Deficiencia de lipasa ácida lisosomal | LIPA |
| Deficiencia de lipoproteinlipasa | LPL |
| Deficiencia de n-acetilglutamato sintetasa | NAGS |
| Deficiencia de ornitina aminotransferasa | OAT |
| Deficiencia de ornitina transcarbamilasa (OTC) | OTC |
| Deficiencia de piruvato carboxilasa | PC |
| Deficiencia de piruvato deshidrogenasa (relacionada con PDHA1) | PDHA1 |
| Deficiencia de piruvato deshidrogenasa (relacionada con PDHB) | PDHB |
| Deficiencia de proteína D-bifuncional | HSD17B4 |
| Deficiencia de tetrahidrobiopterina | PTS |
| Deficiencia de tirosina hidroxilasa | TH |
| Deficiencia de transportador de creatina Ligada al X | SLC6A8 |
| Deficiencia del complejo mayor de histocompatibilidad clase II | CIITA |
| Deficiencia del complejo mitocondrial I / Síndrome de Leigh (relacionado con NDUFAF5) | NDUFAF5 |
| Deficiencia del complejo mitocondrial I / Síndrome de Leigh (relacionado con NDUFAFS6) | NDUFS6 |
| Deficiencia del Factor XI (Hemofilia C) | F11 |
| Deficiencia inmune combinada severa (relacionada con DCLRE1C) | DCLRE1C |
| Deficiencia múltiple de sulfatasa | SUMF1 |
| Deficiencia SAP combinada | PSAP |
| Desorden congénito de la glicosilación (relacionado con ALG6) | ALG6 |
| Desorden congénito de la glicosilación (relacionado con MPI) | MPI |
| Desorden congénito de la glicosilación (relacionados con PMM2) | PMM2 |
| Desorden relacionado con SLC35A3 | SLC35A3 |
| Desórdenes relacionados con DHDDS (incluye Desorden congénito de glicosilación / Retinitis pigmentosa 59) | DHDDS |
| Desórdenes relacionados con MKS1 | MKS1 |
| Desórdenes relacionados con POMGNT1 | POMGNT1 |
| Desórdenes relacionados con PRPS1 (incluye Neuropatía de Charcot-Marie-Tooth tipo 5 y Síndrome de Arts) | PRPS1 |
| Desórdenes relacionados con RPGRIP1L (incluye Síndrome de Joubert 7, Síndrome de COACH y Síndrome de Meckel 5) | RPGRIP1L |
| Desórdenes relacionados con RTEL1 (incluye Disqueratosis congénita) | RTEL1 |
| Desórdenes relacionados con SLC26A2 | SLC26A2 |
| Desórdenes relacionados con WNT10A | WNT10A |
| Diabetes insípida nefrogénica (relacionada con AQP2) | AQP2 |
| Disautonomía familiar | IKBKAP |
| Discinecia ciliar primaria (relacionada con DNAH5) | DNAH5 |
| Discinecia ciliar primaria (relacionada con DNAI1) | DNAI1 |
| Discinecia ciliar primaria (relacionada con DNAI2) | DNAI2 |
| Disferlinopatía (incluye Distrofia muscular de cinturas tipo 2B) | DYSF |
| Disostosis espondilocostal | MESP2 |
| Displasia ectodérmica hipohidrótica | EDA |
| Displasia inmuno ósea de Schimke | SMARCAL1 |
| Distrofia de córnea y sordera de percepción | SLC4A11 |
| Distrofia muscular de cinturas tipo 2A / Calpainopatía | CAPN3 |
| Distrofia muscular de cinturas tipo 2C | SGCG |
| Distrofia muscular de cinturas tipo 2D | SGCA |
| Distrofia muscular de cinturas tipo 2E | SGCB |
| Distrofia muscular de Emery-Dreifuss (relacionado con EMD) | EMD |
| Distrofia muscular relacionada con LAMA2 | LAMA2 |
| Distrofinopatía relacionada con DMD (Incluyendo distrofia muscular Duchenne / Becker y miocardiopatía dilatada) | DMD |
| Encefalopatía Etilmalónica | ETHE1 |
| Encefalopatía neurogastrointestinal mitocondrial (MNGIE) | TYMP |
| Encefalopatía por glicina (relacionada con AMT) | AMT |
| Encefalopatía por glicina (relacionada con GLDC) | GLDC |
| Enfermedad de almacenamiento de glucógeno tipo IB | SLC37A3 |
| Enfermedad de almacenamiento de glucógeno tipo III | AGL |
| Enfermedad de almacenamiento de glucógeno tipo IV / Enfermedad con cuerpos de poliglucosano del adulto | GBE1 |
| Enfermedad de almacenamiento de glucógeno tipo V | PYMG |
| Enfermedad de almacenamiento de glucógeno tipo VII | PFKM |
| Enfermedad de Canavan | ASPA |
| Enfermedad de depósito de ácido siálico libre | SLC17A5 |
| Enfermedad de Fabry | GLA |
| Enfermedad de Gaucher | GBA |
| Enfermedad de Krabbe | GALC |
| Enfermedad de la orina con olor a jarabe de arce (MSUD) tipo 1A | BCKDHA |
| Enfermedad de la orina con olor a jarabe de arce (MSUD) tipo 1B | BCKDHB |
| Enfermedad de la orina con olor a jarabe de arce (MSUD) tipo 2 | DBT |
| Enfermedad de Menkes / Desórdenes relacionados con ATP7A (Síndrome del cuerno occipital y Neuropatía Motora Distal Hereditaria) | ATP7A |
| Enfermedad de Niemann-Pick tipo A / B | SMPD1 |
| Enfermedad de Niemann-Pick tipo C (relacionada con NPC1) | NPC1 |
| Enfermedad de Niemann-Pick tipo C (relacionada con NPC2) | NPC2 |
| Enfermedad de Pompe o de almacenamiento de glucógeno Tipo 2 | GAA |
| Enfermedad de Sandhoff | HEXB |
| Enfermedad de Tay-Sachs / Deficiencia de la hexosaminidasa A | HEXA |
| Enfermedad de Wilson | ATP7B |
| Enfermedad de almacenamiento de glucógeno tipo IA | G6PC |
| Enfermedad granulomatosa crónica (relacionada con CYBA) | CYBA |
| Enfermedad granulomatosa crónica (relacionada con CYBB) | CYBB |
| Enfermedad renal infantil (relacionada con TRMU) | TRMU |
| Enfermedad renal poliquística (relacionada con PKHD1) | PKHD1 |
| Epidermólisis ampollosa de la unión (relacionada con LAMA3) | LAMA3 |
| Epidermólisis ampollosa de la unión (relacionada con LAMB3) | LAMB3 |
| Epidermólisis ampollosa de la unión (relacionada con LAMC2) | LAMC2 |
| Epidermólisis ampollosa distrófica | COL7A1 |
| Fibrosis Quística | CFTR |
| Fiebre mediterránea familiar | MEFV |
| Galactosemia | GALT |
| Galactosemia por deficiencia de galactocinasa | GALK1 |
| Hemocromatosis hereditaria (relacionada con TFR2) | TFR2 |
| Hemocromatosis hereditaria relacionada con HFE | HFE |
| Hemocromatosis hereditaria relacionada con HFE2 | HFE2 |
| Hemoglobinopatías relacionadas con HBB (incluyendo beta-talasemia y enfermedad de células falciformes) | HBB |
| Hipercolesterolemia familiar (relacionada con LDLR) | LDLR |
| Hipercolesterolemia familiar (relacionada con LDLRAP1) | LDLRAP1 |
| Hiperinsulinismo familiar (relacionado con ABCC8) | ABCC8 |
| Hiperinsulinismo familiar (relacionado con KCNJ11) | KCNJ11 |
| Hiperoxaluria primaria tipo 1 | AGXT |
| Hiperoxaluria primaria tipo 2 | GRHPR |
| Hiperoxaluria primaria tipo 3 | HOGA1 |
| Hiperplasia suprarrenal congénita lipoide | STAR |
| Hiperplasia suprarrenal congénita por déficit de 11-beta-hidroxilasa | CYP11B1 |
| Hiperplasia suprarrenal congénita por déficit de 17-alfa-hidroxilasa | CYP17A1 |
| Hiperplasia suprarrenal congénita por déficit de 3-beta-hidroxiesteroide deshidrogenasa, tipo II | HSD3B2 |
| Hipofosfatasia | ALPL |
| Hipoplasia de cartílago-cabello y Displasia Anauxética | RMRP |
| Hipoplasia pontocerebelosa (relacionada con RARS2) | RARS2 |
| Hipoplasia pontocerebelosa (relacionada con SEPSECS) | SEPSECS |
| Hipoplasia pontocerebelosa (relacionada con VRK1) | VRK1 |
| Homocistinuria (relacionada con CBS) | CBS |
| Homocistinuria por deficiencia de MTHFR | MTHFR |
| Homocistinuria, tipo cobalamina E | MTRR |
| Ictiosis congénita (relacionada con TGM1) | TGM1 |
| Inmunodeficiencia combinada grave ligada a X (X-SCID) | IL2RG |
| Inmunodeficiencia combinada severa / Síndrome de Omenn (relacionado con RAG2) | RAG2 |
| Insensibilidad congénita al dolor con anhidrosis | NTRK1 |
| Intolerancia a la fructosa hereditaria | ALDOB |
| Intolerancia a la proteína lisinúrica | SLC7A7 |
| Leucodistrofia metacromática | ARSA |
| Leucoencefalopatía con desvanecimiento de la sustancia blanca (relacionada con EIF2B5) | EIF2B5 |
| Leucoencefalopatía megalencefálica con quistes subcorticales tipo 1 | MLC1 |
| Lipofuscinosis ceroide neuronal (relacionada con CLN5) | CLN5 |
| Lipofuscinosis ceroide neuronal (relacionada con CLN6) | CLN6 |
| Lipofuscinosis ceroide neuronal (relacionada con MFSD8) | MFSD8 |
| Lipofuscinosis ceroide neuronal (relacionada con PPT1) | PPT1 |
| Lipofuscinosis ceroide neuronal (relacionada con TPP1) | TPP1 |
| Lipofuscinosis ceroide neuronal / Epilepsia del Norte (relacionada con CLN8) | CLN8 |
| Lipofuscinosis ceroide neuronal Tipo 3 | CLN3 |
| Microftalmia / Anoftalmía clínica | VSX2 |
| Miopatía miotubular ligada al X | MTM1 |
| Miopatía mitocondrial y anemia sideroblástica 1 | PUS1 |
| Miopatía nemalínica (2) | NEB |
| Miopatía por cuerpos de inclusión 2 | GNE |
| Mucolipidosis tipo II/III (relacionada con GNPTAB) | GNPTAB |
| Mucolipidosis tipo III (relacionada con GNPTG) | GNPTG |
| Mucolipidosis tipo IV | MCOLN1 |
| Mucopolisacaridosis tipo I (incluye los síndromes de Hurler, Hurler-Scheie y Scheie) | IDUA |
| Mucopolisacaridosis tipo II (Síndrome de Hunter) | IDS |
| Mucopolisacaridosis tipo IIIA (Síndrome de Sanfilippo A) | SGSH |
| Mucopolisacaridosis tipo IIIB | NAGLU |
| Mucopolisacaridosis tipo IIIC (Síndrome de Sanfilippo) / Retinitis Pigmentosa 73 | HGSNAT |
| Mucopolisacaridosis tipo IIID (Síndrome de Sanfilippo) | GNS |
| Mucopolisacaridosis tipo IVB (Síndrome de Morquio B) / Gangliosidosis GM1 | GLB1 |
| Mucopolisacaridosis tipo IX | HYAL1 |
| Mucopolisacaridosis tipo VI (Síndrome de Maroteaux-Lamy) | ARSB |
| Neuropatía de Charcot-Marie-Tooth (relacionada con NDRG1) | NDRG1 |
| Neuropatía de Charcot-Marie-Tooth, ligada al X (relacionada con GJB1) | GJB1 |
| Neutropenia congénita (relacionada con HAX1) | HAX1 |
| Neutropenia severa congénita (relacionada con VPS45) | VPS45 |
| Osteopetrosis (relacionada con TCIRG1) | TCIRG1 |
| Paraparesia espástica tipo 15 | ZFYVE26 |
| Paraparesia espástica tipo 49 | TECPR2 |
| Picnodisostosis | CTSK |
| Poliendocrinopatía autoinmune con candidiasis y distrofia ectodérmica | AIRE |
| Polimicrogiria (relacionada con ADGRG1) | ADGRG1 |
| Retinitis Pigmentosa 25 | EYS |
| Retinitis Pigmentosa 26 | CERKL |
| Retinitis Pigmentosa 28 | FAM161A |
| Retinosquisis juvenil ligada al X | RS1 |
| Síndrome alfa-talasemia con retraso mental ligado al X | ATRX |
| Síndrome conos-s / Retinitis Pigmentosa 37 | NR2E3 |
| Síndrome de Aicardi-Goutieres (relacionado con SAMHD1) | SAMHD1 |
| Síndrome de Alport (relacionado con COL4A3) | COL4A3 |
| Síndrome de Alport (relacionado con COL4A4) | COL4A4 |
| Síndrome de Alport ligado al X (relacionado con COL4A5) | COL4A5 |
| Síndrome de Alström | ALMS1 |
| Síndrome de Andermann | SLC12A6 |
| Síndrome de Bardet-Biedl (relacionado con BBS1) | BBS1 |
| Síndrome de Bardet-Biedl (relacionado con BBS10) | BBS10 |
| Síndrome de Bardet-Biedl (relacionado con BBS12) | BBS12 |
| Síndrome de Bardet-Biedl (relacionado con BBS2) | BBS2 |
| Síndrome de Bartter tipo IV | BSND |
| Síndrome de Bernard-Soulier | GP9 |
| Síndrome de Bloom | BLM |
| Síndrome de Carpenter | RAB23 |
| Síndrome de Cockayne tipo A | ERCC8 |
| Síndrome de Cockayne tipo B | ERCC6 |
| Síndrome de Cohen | VPS13B |
| Síndrome de contractura congénita letal 1 / Artrogriposis letal con alteración celular de las astas medulares anteriores | GLE1 |
| Síndrome de depleción del ADN mitocondrial | MPV17 |
| Síndrome de Ehlers-Danlos tipo VIIC | ADAMTS2 |
| Síndrome de Ellis-van Crevels (relacionado con EVC) | EVC |
| Síndrome de Ellis-van Crevels (relacionado con EVC2) | EVC2 |
| Síndrome de Gitelman | SLC12A3 |
| Síndrome de GRACCILE / Desórdenes relacionados con BCS1L (incluye deficiencia de complejo III mitocondrial, Síndrome de Bjornstad, Síndrome de Leigh) | BCS1L |
| Síndrome de Hermansky-Pudlak (relacionado con HPS1) | HPS1 |
| Síndrome de Hermansky-Pudlak (relacionado con HPS3) | HPS3 |
| Síndrome de Joubert 2 / Trastornos relacionados con TMEM216 | TMEM216 |
| Síndrome de Leigh, tipo francocanadiense | LRPPRC |
| Síndrome de Pendred | SLC26A4 |
| Síndrome de Roberts | ESCO2 |
| Síndrome de rotura de Nijmegen | NBN |
| Síndrome de Sjögren-Larsson | ALDH3A2 |
| Síndrome de Smith-Lemli-Opitz | DHCR7 |
| Síndrome de Steel | COL27A1 |
| Síndrome de Stüve-Wiedemann | LIFR |
| Síndrome de Usher Tipo IB / relacionado con el desorden de MYO7A | MYO7A |
| Síndrome de Usher tipo IC (relacionado con el desorden de USH1C) | USH1C |
| Síndrome de Usher tipo ID | CDH23 |
| Síndrome de Usher Tipo IF / relacionado con el desorden de PCDH15 | PCDH15 |
| Síndrome de Usher tipo IIA (relacionado con el desorden de USH2A) | USH2A |
| Síndrome de Usher tipo IIIA | CLRN1 |
| Síndrome de Walker-Warburg / desórdenes relacionados con FKRP | FKRP |
| Síndrome de Walker-Warburg / trastornos relacionados con FKTN | FKTN |
| Síndrome de Zellweger (relacionado con PEX1) | PEX1 |
| Síndrome de Zellweger (relacionado con PEX6) | PEX6 |
| Síndrome de Zellwger (relacionado con PEX10) | PEX10 |
| Síndrome de Zellwger (relacionado con PEX12) | PEX12 |
| Síndrome de Zellwger (relacionado con PEX2) | PEX2 |
| Síndrome del X frágil | FMR1 |
| Síndrome hidroletalus tipo 1 | HYLS1 |
| Síndrome hiperornitinemia-hiperamonemia-homocitrulinuria (HHH) | SLC25A15 |
| Síndrome miasténico congénito (relacionado con CHRNE) | CHRNE |
| Síndrome miasténico congénito (relacionado con RAPSN) | RAPSN |
| Síndrome nefrótico / resistencia a corticoides ( relacionado con NPHS2) | NPHS2 |
| Síndrome nefrótico / tipo finlandés (relacionado con NPHS1) | NPHS1 |
| Sordera autosómica recesiva 77 (DFNB77) | LOXHD1 |
| Sordera y pérdida auditiva no sindrómica (relacionada con GJB2) | GJB2 |
| Tirosinemia tipo I | FAH |
| Tirosinemia tipo II | TAT |
| Trombocitopenia amegacariocítica congénita | MPL |
| Trombofilia relacionada con el Factor V | F5 |
| Trombofilia relacionada con protrombina | F2 |
| Xantomatosis cerebrotendinosa | CYP27A1 |
| Xeroderma pigmentoso, grupo de complementación A | XPA |
| Xeroderma pigmentoso, grupo de complementación C | XPC |
¿A quiénes les recomendamos el estudio?
Considerando que es normal que las personas sean portadoras de al menos una mutación asociada a una enfermedad genética recesiva, le recomendamos el estudio a todos aquellos que estén pensando en tener hijos.
Lo aconsejamos especialmente para:
- Personas con antecedentes familiares.
- Parejas conformadas por personas de una misma etnia con alta frecuencia de enfermedades recesivas.
- Parejas consanguíneas.
- Parejas que desean disminuir el riesgo de tener un hijo con alguna de las enfermedades que se analizan.
¿Querés saber más?
¿Qué son los genes?
El ADN es la molécula en la que se encuentran las instrucciones para fabricar todas las proteínas que los seres humanos necesitan para funcionar correctamente durante toda su vida. También a través del ADN se transmiten las características hereditarias de abuelos a padres y de padres a hijos, como el color de los ojos, la altura, la predisposición a desarrollar enfermedades como la Diabetes o mutaciones asociadas a enfermedades como por ejemplo la Fibrosis Quística.
Cada cadena está formada por otras moléculas llamadas bases.
Las bases son 4:
A, T, C y G (Adenina, Timina, Citosina y Guanina) y forman el alfabeto de la genética. La combinación de esas cuatro letras es la responsable de indicar cómo se fabrican todas las proteínas.
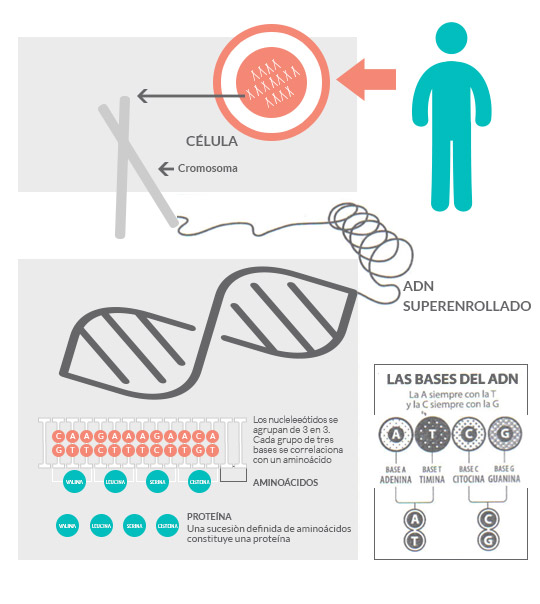
El ADN de los seres humanos se encuentra en el núcleo de cada célula fraccionado en cuarenta y seis unidades independientes, llamadas cromosomas. Estos cuarenta y seis cromosomas pueden dividirse, a su vez, en dos grupos de veintitrés, uno recibido por vía materna y el otro por vía paterna. Esto significa que toda la información genética que llevamos en nuestro ADN está duplicada.
La información genética en los seres humanos

Un gen es un fragmento de ADN que porta toda la información necesaria para la fabricación de una proteína y sus variables.
Por lo tanto, los genes también se encuentran de a pares, es decir están duplicados.
Cuando un gen tiene mutaciones (por ejemplo un cambio puntual, una deleción o una duplicación) puede no funcionar del modo correcto o simplemente no funcionar, siendo esto en muchos casos la causa de una enfermedad.
La mutación del ADN
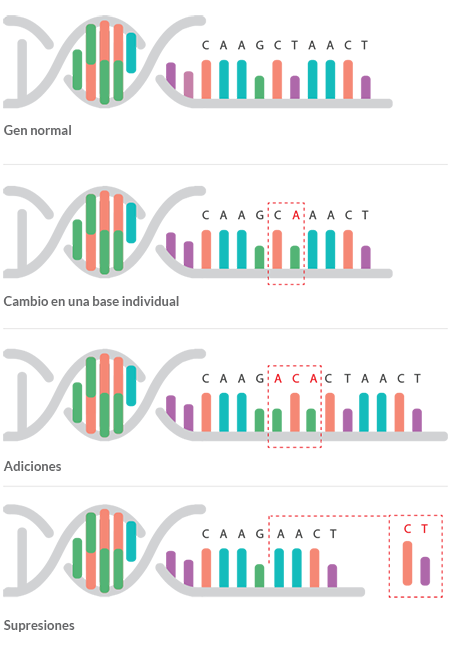
La variación de la secuencia de ADN en un gen puede cambiar la proteína producida por el código genético
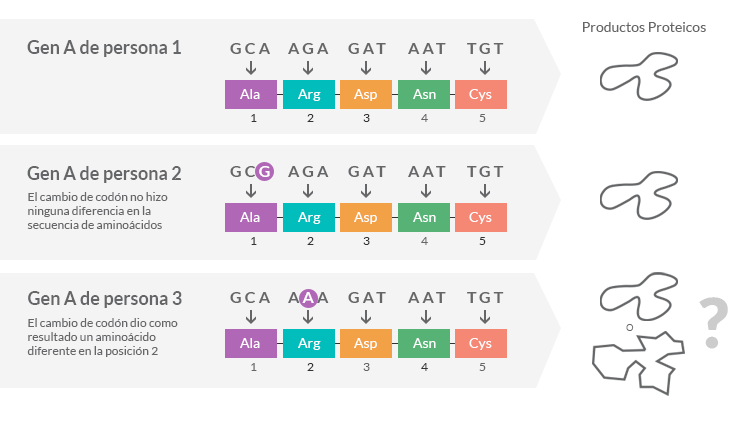
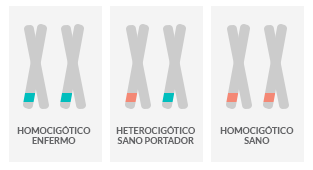
¿Qué es una enfermedad recesiva?
Las enfermedades genéticas recesivas son aquellas que se desencadenan cuando una persona tiene las dos copias (alelos) de un mismo gen alterado (mutado)
Mutaciones Dominantes y Recesivas

¿Qué significa ser portador?
Un individuo que lleva en su genoma una sola copia de un gen alterado (que pudo haberlo recibido de su mamá o de su papá) y que no manifiesta la enfermedad se denomina portador.
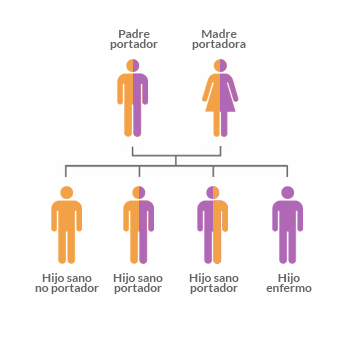
Es normal que las personas sean portadoras de al menos una mutación asociada a una enfermedad genética recesiva. Por lo tanto estas mutaciones o copias de genes alterados, se transmiten silenciosamente dentro de una familia de generación en generación y, en muchos casos, sólo llegan a descubrirse cuando una pareja tiene un hijo que la manifiesta.
¿Por qué es importante saber si las dos personas de una pareja son portadoras?
Si dos personas son portadoras de un mismo gen alterado tienen un 25% de probabilidades de tener un hijo afectado para esa enfermedad.
Si sólo un miembro de la pareja es portador de una copia de un gen alterado para una enfermedad recesiva, la probabilidad de que el hijo sea portador es de un 50% y la probabilidad de que ese hijo no presente síntomas para esa enfermedad es del 100%.
¿Qué es un screening?
Un screening preconcepcional es un estudio que permite analizar si una persona es portadora de alguna mutación para un número definido de enfermedades recesivas.
Si se analiza a ambos miembros de la pareja pueden evaluarse las probabilidades de que esa pareja tenga un hijo que manifieste alguna de las enfermedades analizadas.
GendaFamily analiza si una persona es portadora de mutaciones para un gran número de enfermedades genéticas recesivas. Si los resultados indican que las dos personas de una pareja tienen una mutación para la misma enfermedad, podrán discutir con su médico genetista las distintas alternativas para planificar el embarazo.
Las alternativas posibles son, por ejemplo, realizar un estudio preimplantatorio o estudios prenatales, adoptar o considerar la donación de óvulo o esperma. Tomar una decisión deberá depender también de la categoría en la que está clasificada la enfermedad recesiva identificada en el estudio.
Categorías de enfermedades recesivas analizadas:
- Enfermedades con tratamiento limitado o sin tratamiento.
- Enfermedades asociadas a baja expectativa de vida.
- Enfermedades asociadas a discapacidad intelectual.
- Enfermedades cuyos síntomas pueden mejorar con intervención temprana.
